Para entender este post, antes leia o post sobre a Doença de Huntington.
O que são doenças de Huntington-like? São doenças que se parecem com a doença de Huntington (DH), mas obviamente não são. O diagnóstico é afastado por conta da ausência da mutação característica da DH, no braço curto do cromossomo 4 (observe abaixo).
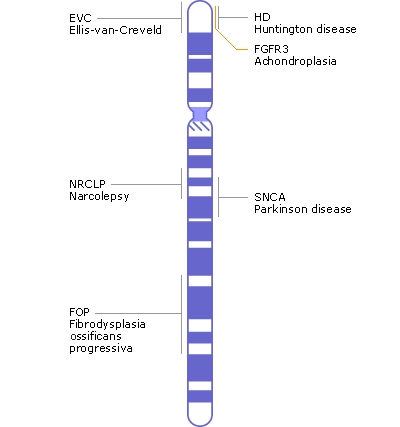 |
| http://www.news-medical.net/image.axd?picture=2010%2F4%2Fa04chr.jpg |
Acima encontramos um esquema do cromossomo 4, com seus dois braços, o curto (acima) e o longo (abaixo). Todo cromossomo tem seu braço curto e longo. Vemos que há vários genes que produzem várias doenças diferentes, e que se encontram neste cromossomo. E o local da mutação da DH está no extremo do braço curto (lá em cima). Com testes genéticos específicos, é possível dar o diagnóstico através do achado desta mutação. Quando há o fenótipo, ou seja, a clínica de DH, mas a mutação não é encontrada (e a DH é, talvez, a única doença genética neurológica que é causada por uma única mutação), dizemos que o paciente possui uma doença de Huntington-like, ou DHL. Há 4 formas clássicas de DHL, numeradas de 1 a 4, mas várias outras doenças podem simular a DH, e serão somente comentadas aqui.
A DH é rara, mas as DHL são ainda mais raras, sendo que dos casos de DH estima-se que 1% a 7% sejam DHL. Os casos de DHL podem ser classicamente parecidos com a DH, ou seja, coreia com alterações comportamentais e mentais, ou podem apresentar quadros diferentes, como uma síndrome parkinsoniana (ou seja, que se parece com a doença de Parkinson, com lentidão, rigidez e tremor de repouso), ou uma síndrome de contração muscular involuntária mantida, ou seja, uma síndrome distônica. Pode acometer adultos ou jovens, e mais raramente crianças.
As DHL's podem ocorrer em qualquer lugar do mundo, mas há exceções. A DHL2 é achada quase que exclusivamente em pacientes de ancestralidade sulafricana (com exceção de uma única família brasileira de ascendência portuguesa e espanhola, ou seja, caucasiana). A mutação aqui é bem diferente da da DH (ocorre em um gene que codifica uma proteína, chamada de junctofilina-3).
A DHL2 é muito parecida com a DH, inclusive na idade de início e nas características clínicas como coreia. Inclusive as alterações na ressonância magnética (veja no post anterior sobre DH) são muito semelhantes às alterações causadas pela DH.
Em pacientes japoneses, há uma doença rara (e que, apesar de ser quase que exclusiva de pacientes japoneses, foi descrita recentemente também no Brasil em uma família de caucasianos), chamada de atrofia dentato-rubro-pálido-luysiana (DRPLA) (Ufa!). Há várias outras doenças que se parecem com a DH, e que podem ser classificadas como DHL.
Há outro grupo raro de doençças (e que eu já vi em três pacientes jovens) que são as síndromes de neuroacantocitose, onde há manifestações neurológicas, como coreia e movimentos involuntários da boca e língua, com alterações comportamentais e mentais e alterações das células vermelhas do sangue, que ficam cheias de espículas, virando acantócitos. Há duas doenças clássicas deste grupo, a córeo-acantocitose e a síndrome de McLeod.
Há outra doença, mais comum em crianças e jovens, que aparece como dificuldade progressiva da marcha e da fala, com distonia a parkinsonismo, que é a degeneração relacionada à pantotenato-kinase, ou PKAN (antigamente chamada de doença de Hallervorden-Spatz). E uma doença tratável e que pode se assemelhar, pelo menos inicialmente, à DH é a Doença de Wilson, que se caracteriza por depósitos de cobre no corpo (leia sobre ela aqui).
Bem, há doenças que se parecem com a DH, mas não são a DH, como já visto. E entre elas, a mais recentemente descrita é a DHL4, que na verdade é uma ataxia, a ataxia espinocerebelar tipo 17 (há mais de 30 tipos de ataxias espinocerebelares, ou SCA's). Mas o que é ataxia? Veja um breve comentário sobre isso aqui. A SCA17 relaciona-se a outro gene completamente diferente da DH, e se caracteriza mais pela ataxia cerebelar, mas pode haver distonia e coreia, como na DH.
A HDL1 é uma doença priônica (doença priônica??? Bem, leia sobre isso aqui). Uma característica dessa doença é a presença de crises epilépticas, algo que não ocorre na DH, ou se ocorre, o faz raramente. A HDL1 é muito, mas muito rara, foi relatada em somente uma família até agora, e relaciona-se à mutação de uma proteína que todos nós temos, a proteína priônica, ou PRNP, que é decodificada a partir do gene da proteína priônica. Não tem nada a ver com a famosa doença da vaca louca, ou a doença de Creutzfekdt-Jacob.
Já a DHL3 ocorre em crianças, e foi descrita em duas famílias da Arábia Saudita. De acordo com o Genetics Home Reference (base de dados genética da US National Library of Medicine), a DHL3 provavelmente não será mais incluída como DHL por conta de diferentes padrões genéticos e clínicos.
De todas as DHL's a mais comum é a DHL4, e depois é a DHL2.
E como fazer o diagnóstico?
Primeiro, claro, há a necessidade de um neurologista que conheça as doenças mencionadas acima e seja especialista na área. O diagnóstico começa com a história e o exame neurológico.
Exames de sangue, quando indicados, podem ser solicitados pelo médico. O diagnóstico da doença de Wilson, com a medida da ceruloplasmina e do cobre na urina de 24 horas, além do achado do anel de Kayser-Fleischer (veja no post sobre doença de Wilson, referido acima), deve ser feito nos pacientes com suspeita da doença, pois é condição tratável.
Exames de imagem, como a ressonância magnética, devem ser solicitados de acordo com indicação clínica, e podem servir como meio de diagnosticar certas doenças que se parecem com a DH.
Já os testes genéticos só devem ser solicitados sob orientação do médico que assiste o paciente.